赖文珍研究方向
课题组研究方向
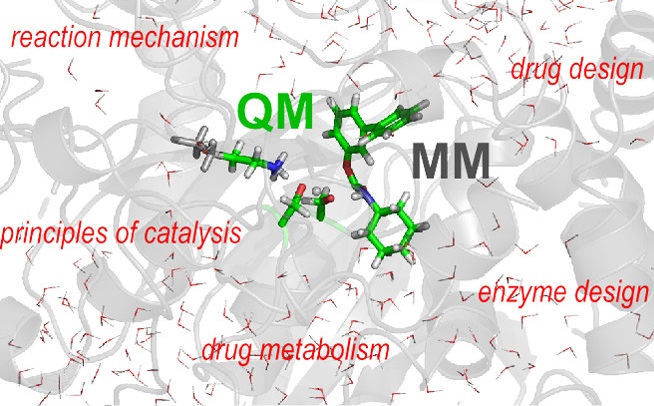
1.酶促反应机理的理论计算——使用QM/MM方法
混合量子力学/分子力学(QM/MM)模拟已经成为研究生物化学反应的一种流行工具。在QM/MM方法中,化学过程发生的系统区域在量子化学理论的适当水平上进行处理,而其余部分则由分子力学力场描述。
2. 小分子催化机理的理论计算——使用DFT方法
高斯16提供了各种各样的密度泛函理论(DFT)模型。能量、解析梯度和真解析频率可用于所有DFT模型。使用DFT计算通过高斯可以确定最佳的几何形状,自由能和化学位移为每个构象。这对我们了解许多小分子的各种性质是非常有用的。
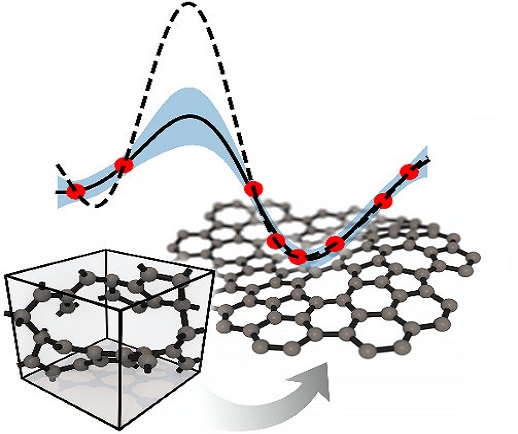
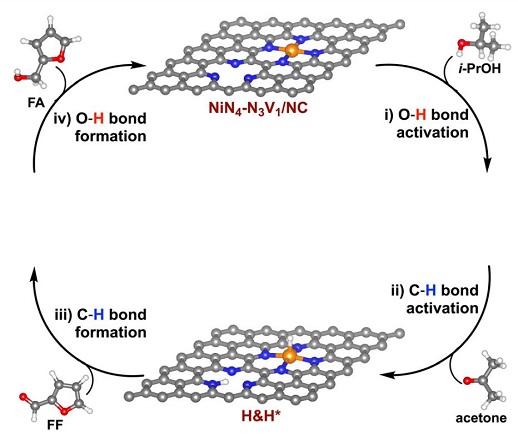
3. 石墨烯表面催化反应机理的理论计算——使用第一性原理方法
计算材料科学侧重于建立可预测或可描述的模型,以帮助研究材料的潜在机制,减少开发新材料的时间和成本。VASP是目前在材料微观反应机理科学研究和材料电子结构性能计算中应用最为广泛的软件。它可以处理金属和氧化物、半导体、晶体、掺杂系统、纳米材料、分子、簇、表面系统和界面系统